Computational Chemistry · Teaching Note
讀懂 Nico 的 Cas9 變體篩選漏斗(v5 / v6)
用物理人的語言,把每一層在幹嘛、Nico 想往哪走、以及 kinetics 那一維該怎麼談,一次講清楚。
🕓 怎麼看更新:§0–§8 教材本體為 2026-07-04 初版;之後隨計算進度補的章節/區塊都掛了一個 +日期 標記,代表該內容的新增/更新時間。沒掛標記的即為初版內容。
用物理人的語言,把每一層在幹嘛、Nico 想往哪走、以及 kinetics 那一維該怎麼談,一次講清楚。
🕓 怎麼看更新:§0–§8 教材本體為 2026-07-04 初版;之後隨計算進度補的章節/區塊都掛了一個 +日期 標記,代表該內容的新增/更新時間。沒掛標記的即為初版內容。
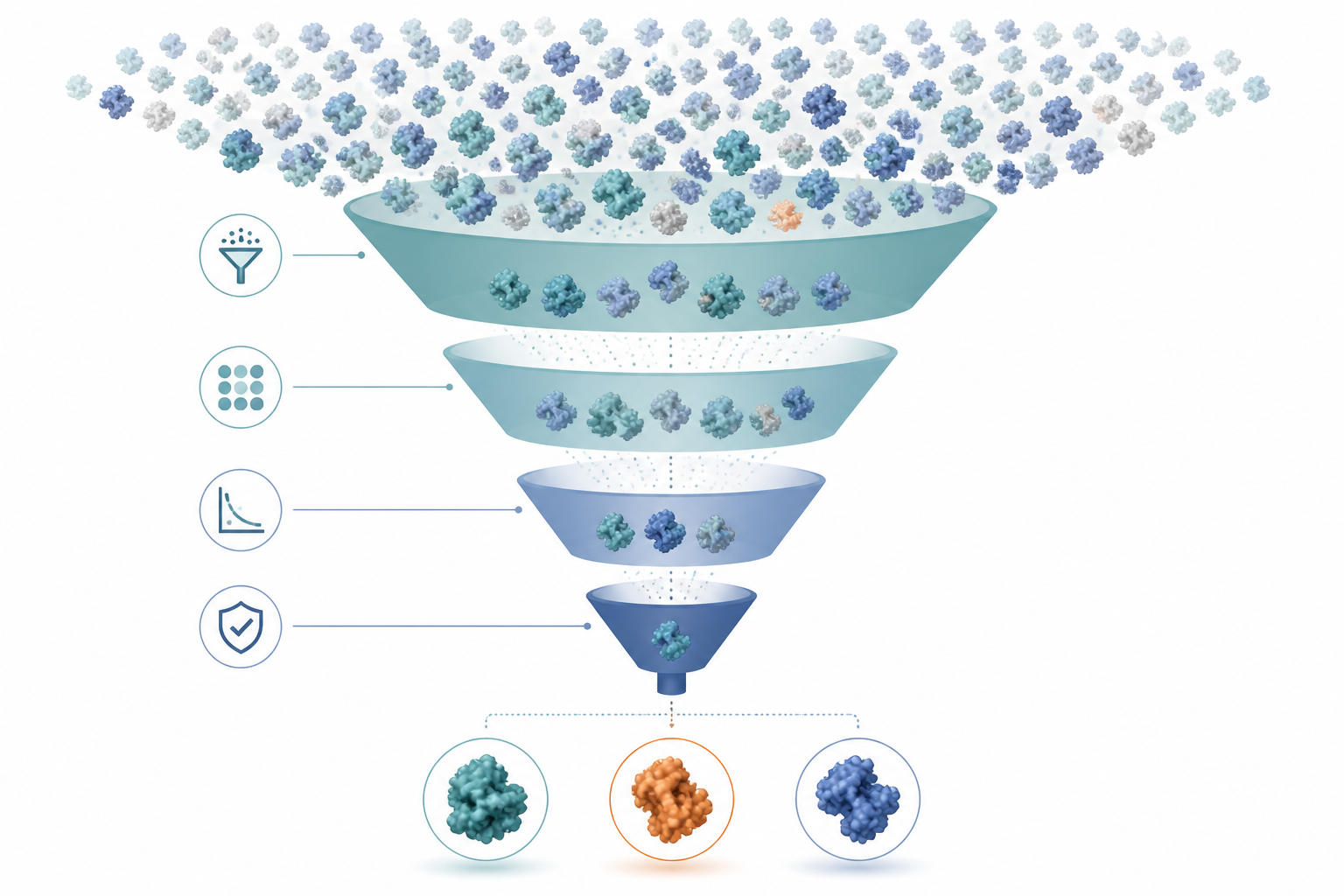
Nico 這台漏斗,本質是候選削減機,不是「物理真理機」。SpCas9(1368 殘基)的多點突變庫組合爆炸(10⁴–10⁵⁺),濕實驗一輪只做得起 ~10–50 個,所以用便宜、會看核酸、可以有雜訊的篩子在頂層把庫富集,把貴的物理留給少數存活者。
| 層 | 處理量 | 在算什麼 | 你的類比 |
|---|---|---|---|
| Phase 1 序列 | 10⁴–10⁵ | 序列合不合理(幾何/親和/演化/摺疊/溶解) | 便宜 descriptor 初篩 |
| Phase 2 結構 | 100–500 摺疊 | 疊出 all-atom 複合體 + 清座標 | 建結構 + minimize |
| Phase 3 打分 | 10–100 | 結合能 ΔGbind(三條路) | 隱溶劑單點 / AIMD 系綜 |
| Phase 4 FEP | 1–5 | 精確相對 ΔΔGbind | RPA / CCSD(T) 級把關 |
為什麼需要漏斗:20 個位點就有 ~10⁵ 雙突變、~10⁷ 三突變,濕實驗做不起百萬個。漏斗唯一的任務:從天文數字的庫裡便宜地挑出「值得真的合成」的 ~10–50 個。
三個設計直覺(讀懂 Nico 每個選擇的鑰匙):
全部「只吃序列、不折疊、一次前向推論」。六個正交閘門:
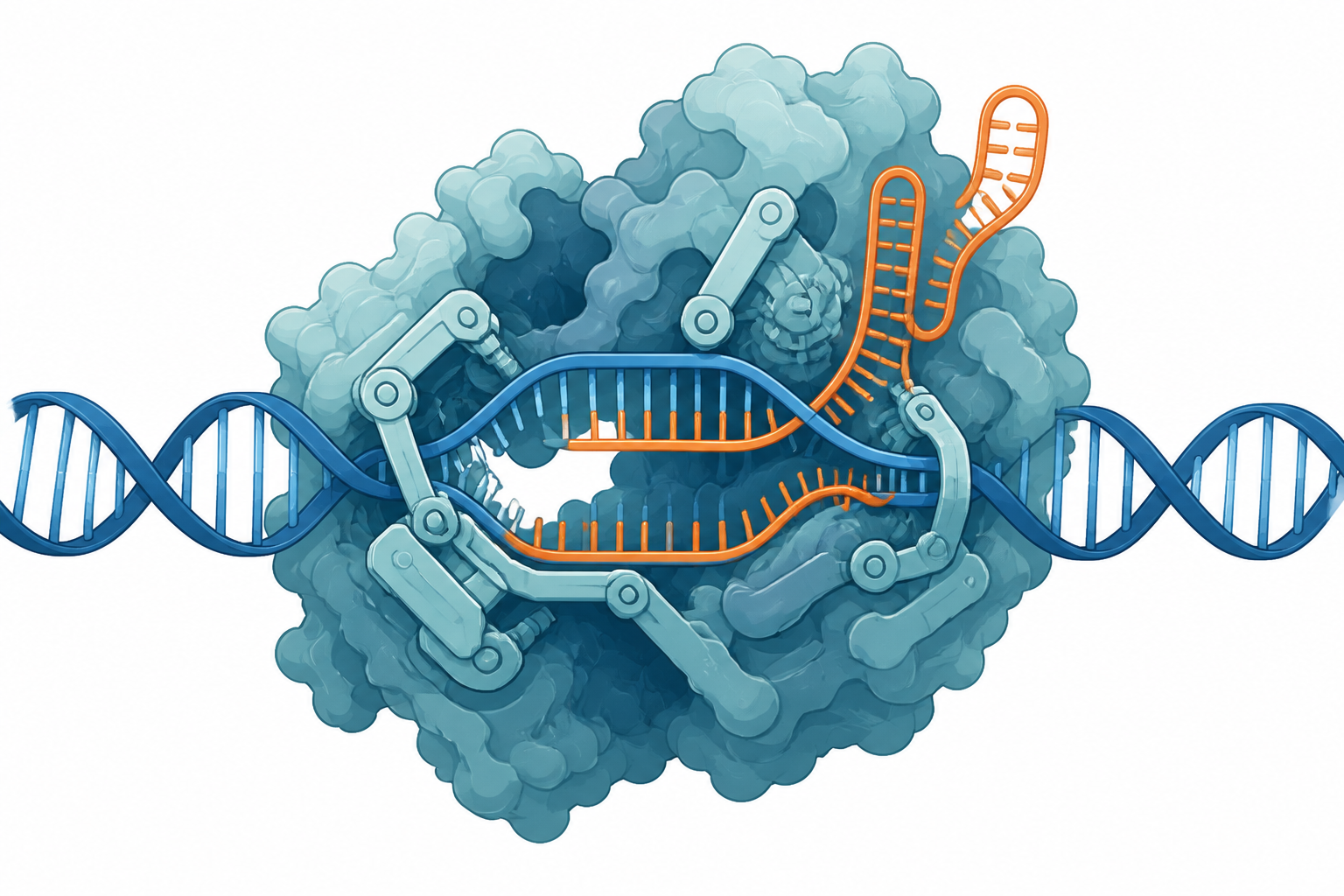
為什麼需要 3D:Phase 1 全是「序列進、分數出」,看不到「這顆突變側鏈實際擺哪、跟哪個 DNA 磷酸撞到」。類比:光有化學式 Fe₃Ni 排不出能量,得先有 POSCAR。
概念(你會很有共鳴):沿一個非物理的 λ 座標把 WT 側鏈煉金術般 morph 成 mutant,在複合體與 apo 各做一次,靠熱力學循環讓算不出的邊抵消,得到精確 ΔΔGbind = ΔGmut(複合體) − ΔGmut(apo)。pmx 建 hybrid 拓撲,MBAR 把所有 λ 窗最優合成一個數+誤差(=WHAM 的 binless 繼承者)。
物理類比:就是你認得的熱力學循環(Hess/Born-Haber)+ TI / adiabatic switching。
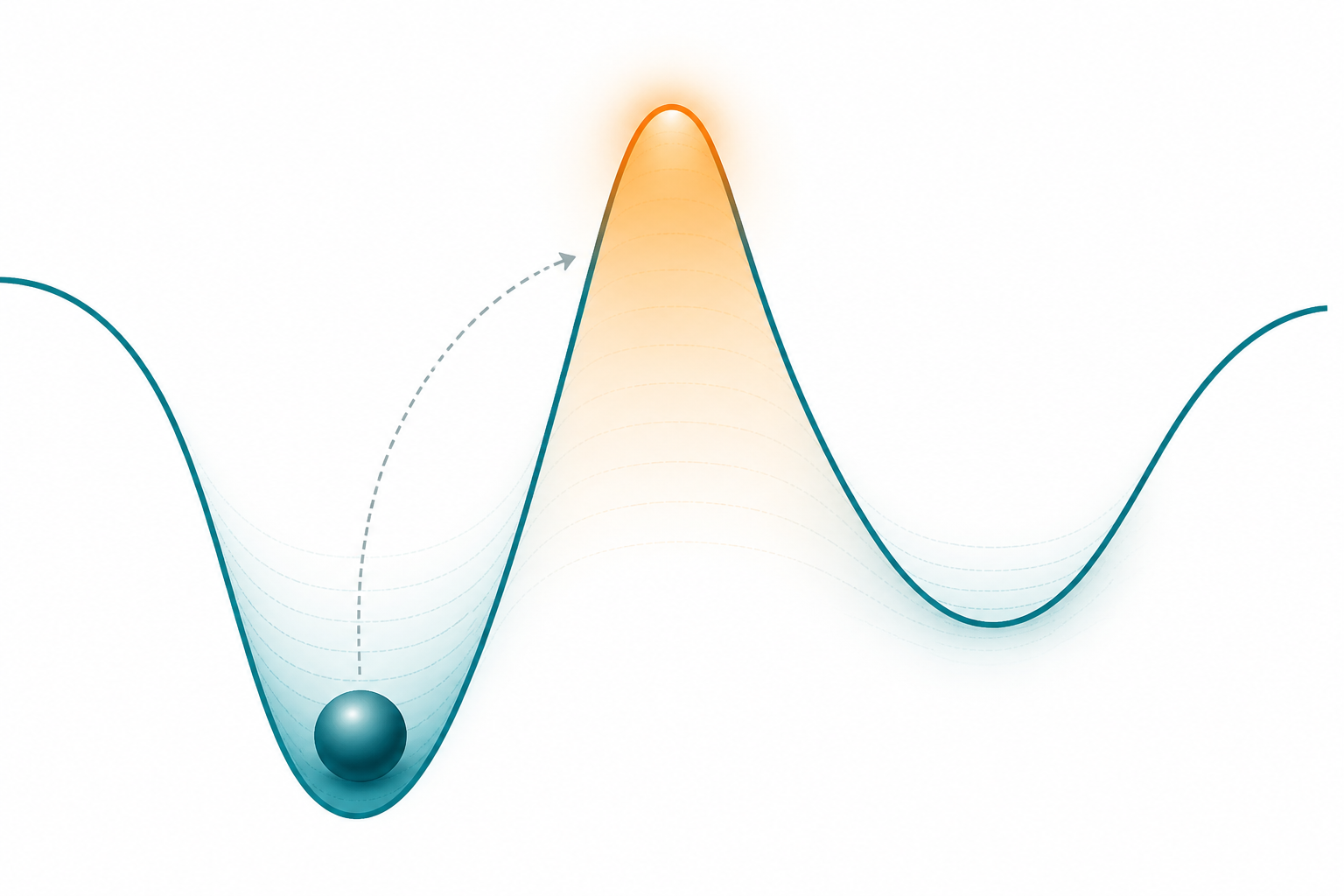
⚠ 範圍是蛋白胺基酸突變(不做 DNA/RNA 鹼基 alchemy)。而且它是 end-state 平衡量:再精確,對速率、障礙、構形檢查點一無所知。
| 版本 | 動作 | 透露的意圖 |
|---|---|---|
| v3/v4 | 通用工具(ESM-IF1)+ 有 off-target specificity MD tier | 泛用 + 有嘗試碰專一性 |
| v5 | 換核酸感知工具、模組化打分、AF3 primary;拿掉 specificity tier;DL 推論當 primary | 「現代化、快、模組化」——但唯一碰專一性的層消失了 |
| v6 | 加 thermoMPNN;Gate 2b 做成 FoldX/Mini-MD 選擇;把 靜態 MM-PBSA 升為 primary | 預設越來越物理——把物理邊界往漏斗上方推 |
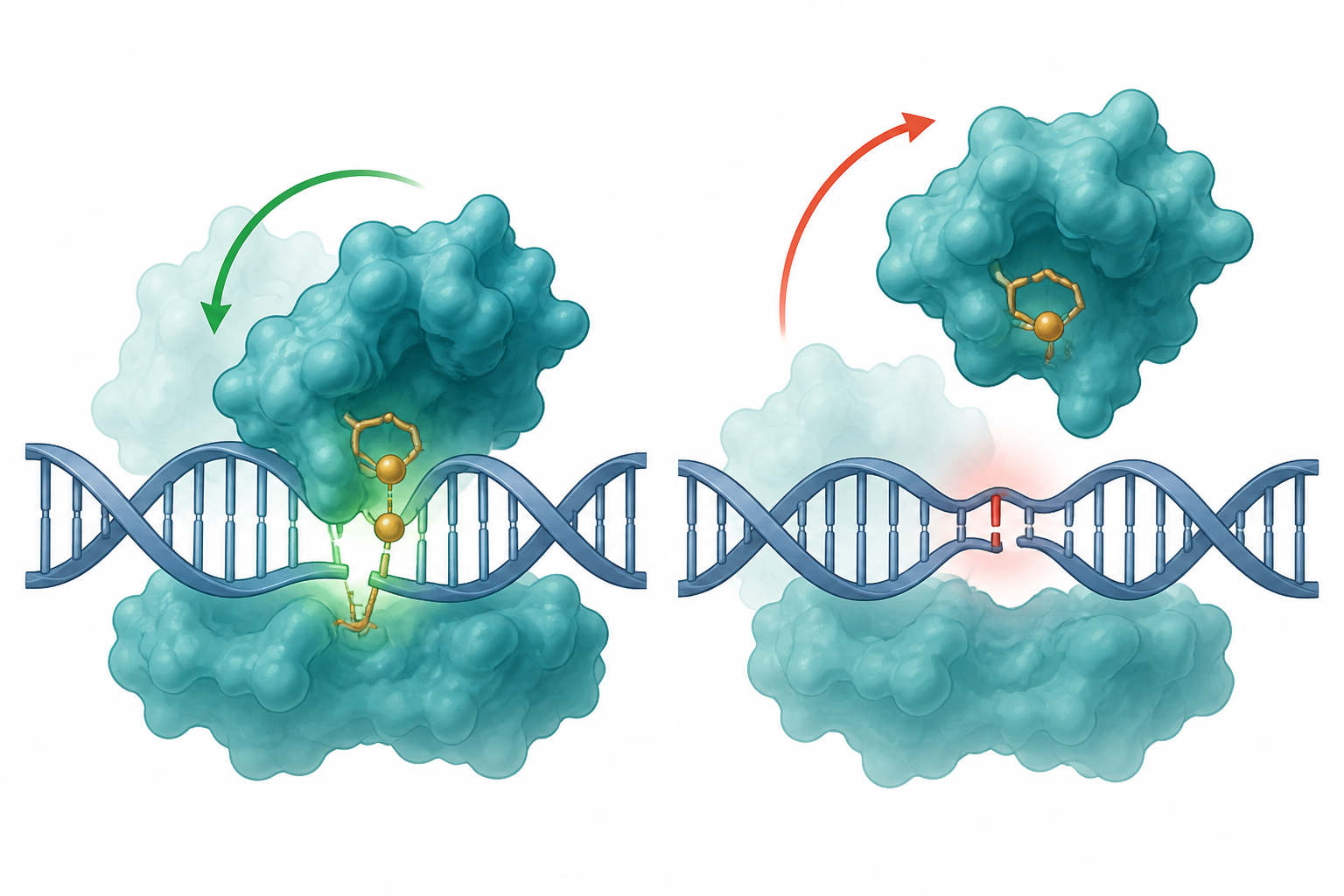
Cas9 不是「結合=切」的開關,是多步機器。辨識 on/off-target 主要發生在第 3 步(構形檢查點),不是結合那步:
前面都在講「該怎麼讀這台漏斗」。這一節是進度:漏斗上游的幾個閘門,我們已經在國網 Nano5 GPU 上實際跑起來了。更關鍵的是——Gate 2a 摺出來的結構,剛好給了上一節 kinetics 論點一個我們自己算出來的證據。




相關上線頁:OpenMM GPU MD demo · 為什麼結合自由能漏斗量錯了東西(kinetics 論點)。所有圖為國網 Nano5 實算輸出,非示意。
§★ 的 apo MD 有一句誠實 caveat:「這是沒放核酸的單獨蛋白,完整 RNP 會束縛部分運動」。這一節就是去補它——把 sgRNA + 目標 DNA 放回去,跑真正的三元複合體(Cas9 + RNA + DNA),在國網 Nano5 H100 上跑了 8 條、每條約 93.5 ns 的軌跡。結論先講:HNH 綁上完整 RNP 之後,仍然是最會動的域——這是指向「HNH=動力學檢查點」的又一條、也是最完整的證據線(完整複合體實測)。
一句話:整台機器切不切、切得準不準,取決於 HNH 這把剪刀能不能擺到定位——而「能不能擺、擺多快」是動力學,不是結合強度。所以我們一直在問:MD 裡 HNH 是不是真的特別會動?
↗ 看靜態互動版(3D+4-replica 疊圖) ‧ ↗ 看全螢幕動畫版
這一段把每個設定攤開講,因為對你這種 MD 背景的人,「參數對不對」比「結果漂不漂亮」更該先看。整體就是一套標準的顯式溶劑生產 MD,只是分子大、且要處理核酸端點。
| 設定 | 值 | 物理類比 / 為什麼 |
|---|---|---|
| 起始結構 | Chai-1 預測的 RNP(不是晶體) | 要「現在就能上機」+ AI 模型完整含 R-loop。代價:介面偏鬆(AI 摺的接觸不夠緊)→ 只有大效應能冒出雜訊。 |
| 核酸 5′ 端處理 | 剝掉首殘基 P/OP1/OP2 → 5′-OH | amber14 沒有「5′-磷酸端」的 residue template。就像 VASP 要選對 POTCAR——力場每個端點都要有對應模板,否則 OpenMM 直接報 template not found。 |
| 盒子 / 溶劑 | 14.22 nm 立方、272,791 atoms(WT;eSpCas9 273,014)、TIP3P 顯式水 | 標準週期性水盒。原子數大是因為完整複合體+足夠水層。 |
| 離子 | 0.15 M NaCl(中和+生理離子強度) | 核酸帶大量負電,必須中和;0.15 M 對齊生理條件。 |
| 力場 | amber14 = ff14SB(蛋白)+ OL15(DNA)+ OL3(RNA) | 就像 DFT 選 functional:蛋白/DNA/RNA 各有各的參數集,要相容。這組是核酸 MD 的主流選擇。 |
| 平衡 | restrained 多階段(Skeens 式) | 先 minimize(對溶質重原子加 k=300 kcal/mol/Ų 諧振約束);再分段升溫 0→100→200→300 K,約束 100→25→5→0 逐步鬆綁;最後 ~5 ns 定密度。就是你生產 MD 前的 minimize+equilibrate,只是對「AI 摺的鬆結構」加位置約束逐步釋放,避免一放開就炸。 |
| 生產 | 300 K、NPT(MonteCarlo barostat 1 bar)、Langevin 恆溫 | 標準有限溫度系綜。NPT 讓密度自洽。 |
| 時間步 | 4 fs + HMR(氫質量重分配 ×1.5) | HMR 把氫的質量勻一點給鄰接重原子、放慢高頻 X–H 振動,讓時間步能拉大(保留平衡熱力學,只放慢含氫模態)=近乎免費加速。⚠ 誠實 flag:這裡 H 質量只設 1.5 amu,對 4 fs 偏輕(慣例 ~4 amu 才配 4 fs;apo run 同樣 1.5 amu 只敢用 2 fs)。實測穩定(RMSD 收斂 ~2.7 Å)故先導可接受,但這步偏積極,嚴格版建議 H 用 ~3–4 amu。 |
| replica | 4 條,seed = 1/2/3/4 | seed 餵初始速度(Maxwell–Boltzmann 抽)+ Langevin 隨機力+ barostat。MD 是混沌的,起始差一點點幾 ps 後就發散 → 4 條 = 4 次獨立抽樣 → 誤差棒。判斷突變效應真偽要「Δ 大於 replica 散布」。 |
| 長度 / 算力 | 每條 ~93.5 ns(4678 幀 × 20 ps) | 目標本是 100 ns,被 SLURM wall-time 切在 93.5(可 checkpoint 續跑)。8 條共 ~87 H100-GPU-小時,速度 ~207 ns/day。 |
把 4 條軌跡的逐殘基 Cα RMSF(每個殘基在軌跡裡晃多大)平均起來、再按結構域分組,客觀排下來:

光看 WT 還不夠。我們想知道:這套 pipeline 有沒有能力「看到」一個已知的突變效應?於是拿現成的高保真變體 eSpCas9(1.1)(三個突變 K848A / K1003A / R1060A,中和抓住位移股的正電凹槽)跑同一套流程、4 條 replica,算 Δ(eSpCas9 − WT)。預期:eSpCas9 應該讓 non-target 股更鬆、RMSF 更高。

本節所有數據為國網 Nano5 H100 上 8 條 ~93.5 ns 三元 MD 的實算輸出(非示意)。互動 3D 由 3Dmol.js 繪製。
§★ 是「我們已經跑的上游閘門」(apo 蛋白)。這一節是 Nico 剛傳來的兩份文件——一份 07-07 會議整理,一份給你的 MD benchmark protocol(對齊 Skeens 2024)。一句話:在拿 MD 去排「未知的」設計變體之前,先用它重現「已知答案」的系統,確認參數是對的。這正是你熟的——選 functional 前先算已知 lattice constant / formation energy 對標。
不能一開始就用 MD 去排你不知道答案的新變體——你怎麼知道它算得準?protocol 的作法是:先跑一批文獻上答案很清楚的 Cas9 專一性變體(eSpCas9 / HF1 / HypaCas9 / evoCas9),它們都有已發表的 MD + 實驗結果。如果我們的 MD 能重現這些已知趨勢 → 參數可信 → 才拿去算新的設計變體。
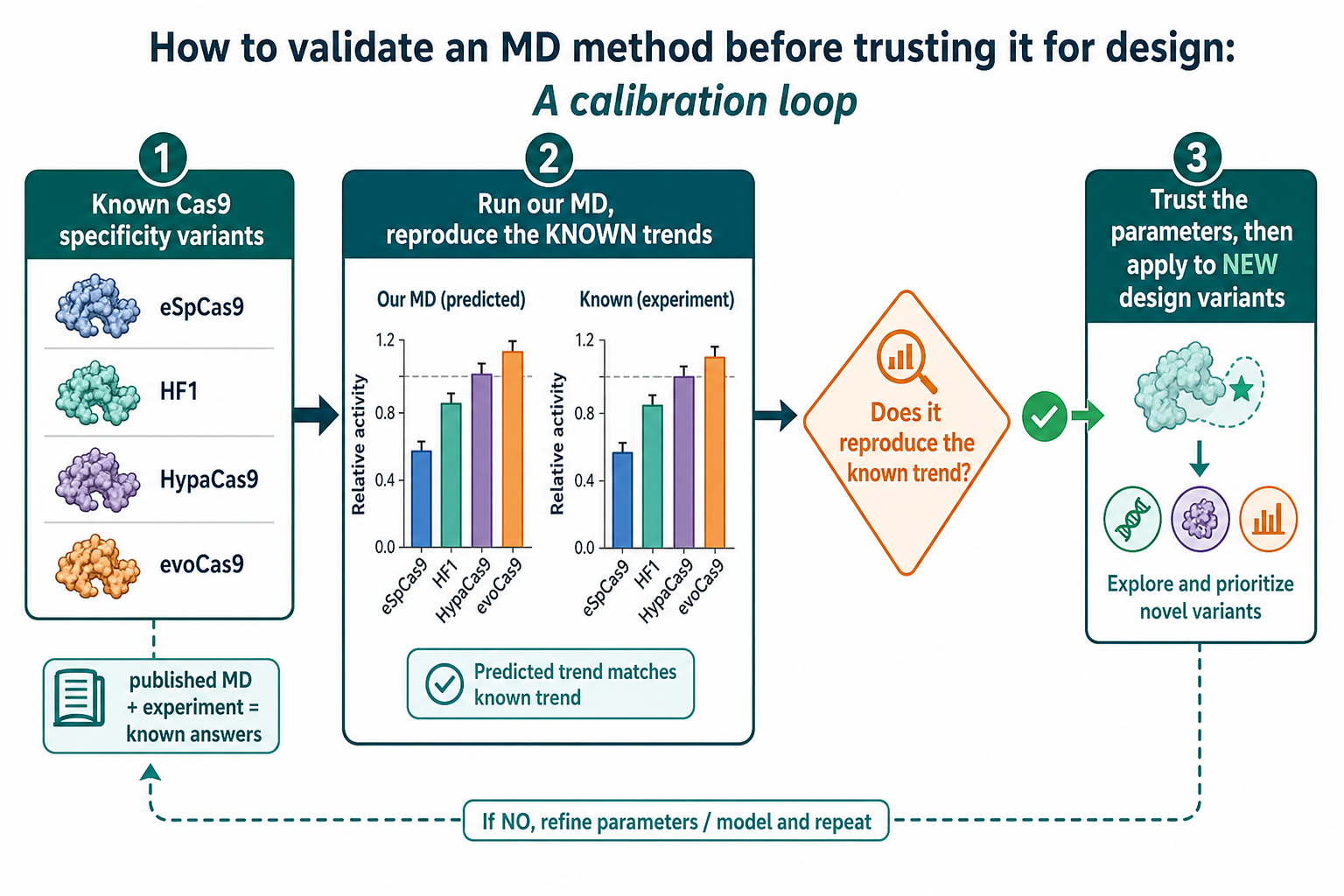
protocol 一開頭就把要量的東西切成兩條軸,而且明說要分清楚:
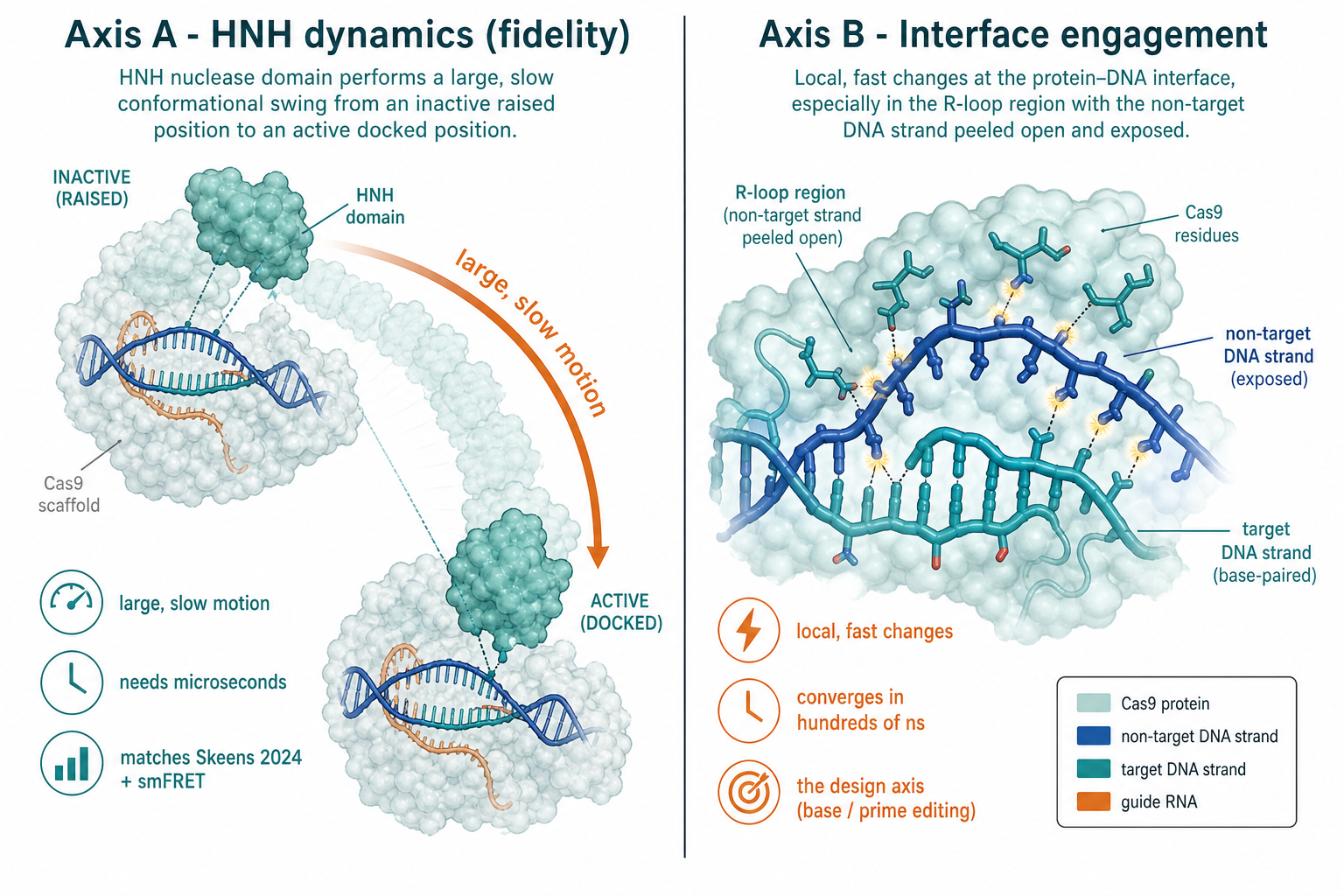
| 軸 | 量什麼 | 快慢 | 對這專案 |
|---|---|---|---|
| Axis A HNH 動力學 | HNH 大幅構形擺動、allosteric network(對齊 Skeens 2024 + smFRET) | 慢:multi-µs(Skeens ~16 µs/系統) | 只當參數校準;你的預算通常到不了 |
| Axis B 介面 engagement | R-loop 穩定度、non-target 股外露、介面接觸 | 快:~數百 ns 收斂 | 設計真正要的軸(BE / PE engagement) |
protocol 的核心是一張對標突變清單:每個系統都有明確功能、已發表的 MD/實驗、和要看的觀測量。FAST=介面 / 局部,數百 ns 可得;SLOW=HNH allostery,要 multi-µs。
| 系統 | 突變(域) | 機制/要驗的(文獻) | 快慢 |
|---|---|---|---|
| WT | — | 基準:複合體穩定度、R-loop 完整、HNH 構形(Palermo 2017 / Skeens 2024 / smFRET) | both |
| eSpCas9 | K848A / K1003A / R1060A(non-target 股溝槽) | 中和穩定被頂開 non-target 股的正電溝槽 → 去穩 off-target R-loop(Slaymaker 2016) | FAST · BE 相關 |
| SpCas9-HF1 | N497A / R661A / Q695A / Q926A(Rec3+HNH 旁) | 移除對 target 股骨架的氫鍵 → 去掉「多餘結合能」→ 較不容忍 mismatch(Kleinstiver 2016) | FAST |
| HypaCas9 | N692A / M694A / Q695A / H698A(Rec3 α37) | Rec3 / α37 突變 → allosteric 降低 HNH 柔性 → 增強 proofreading(Chen 2017) | SLOW |
| evoCas9 | M495V / Y515N / K526E / R661Q(Rec3) | 高專一性但 HNH 動力學 ≈ WT(不同機制)→ 當鑑別對照(Casini 2018) | SLOW |
| WT + PAM-distal mismatch | DNA 端 protospacer 18–20 錯配(非蛋白突變) | off-target R-loop 去穩 → 你的 Δ(on−off) engagement 軸(Singh 2016) | FAST-ish |
| dCas9 | D10A + H840A(催化死) | 結合 / 形 R-loop 如 WT,只是不切 → 穩定度對照 | FAST |
建議跑序(protocol §6,先做便宜 FAST 的):① WT(必須先過)→ ② eSpCas9 & HF1(介面 benchmark)→ ③ WT+mismatch(Δ on−off)→ ④ dCas9 / nickase(穩定度對照)→(預算到 µs 才做)⑤ HypaCas9 + evoCas9(重現 Skeens 的 HNH 柔性模式)。
🕓 這一節是滾動更新的進度:標有 +日期 的區塊代表該內容的新增/更新日期,方便看出哪些是後來補的。
§★ 那條 MD 是 apo 蛋白(沒核酸)。現在照這份 protocol,我已經把 WT 三元複合體(system #1,正對照)建好、在國網 Nano5 上跑起來了:
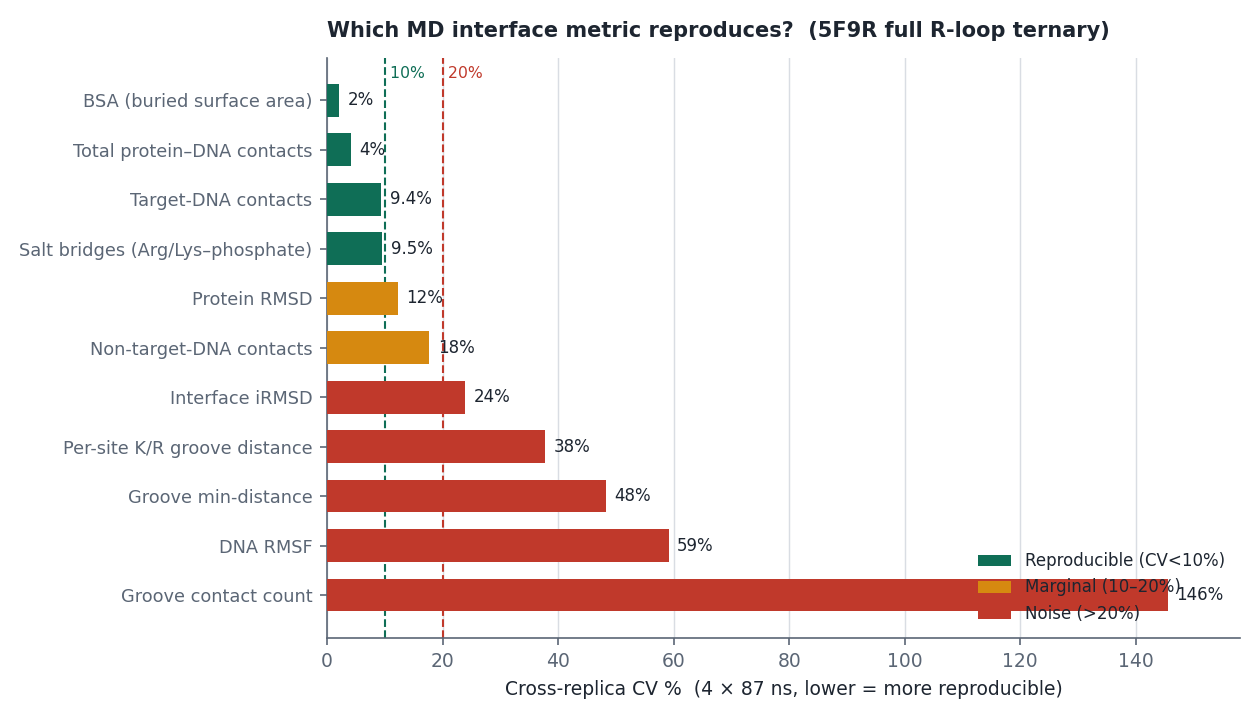
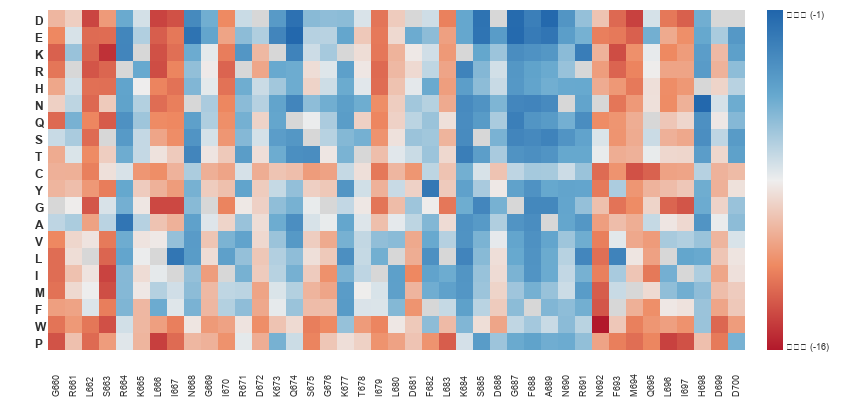
mean_contacts。ARG765(rep1)跑完 = 28.66(平均每幀約 29 對貼近)。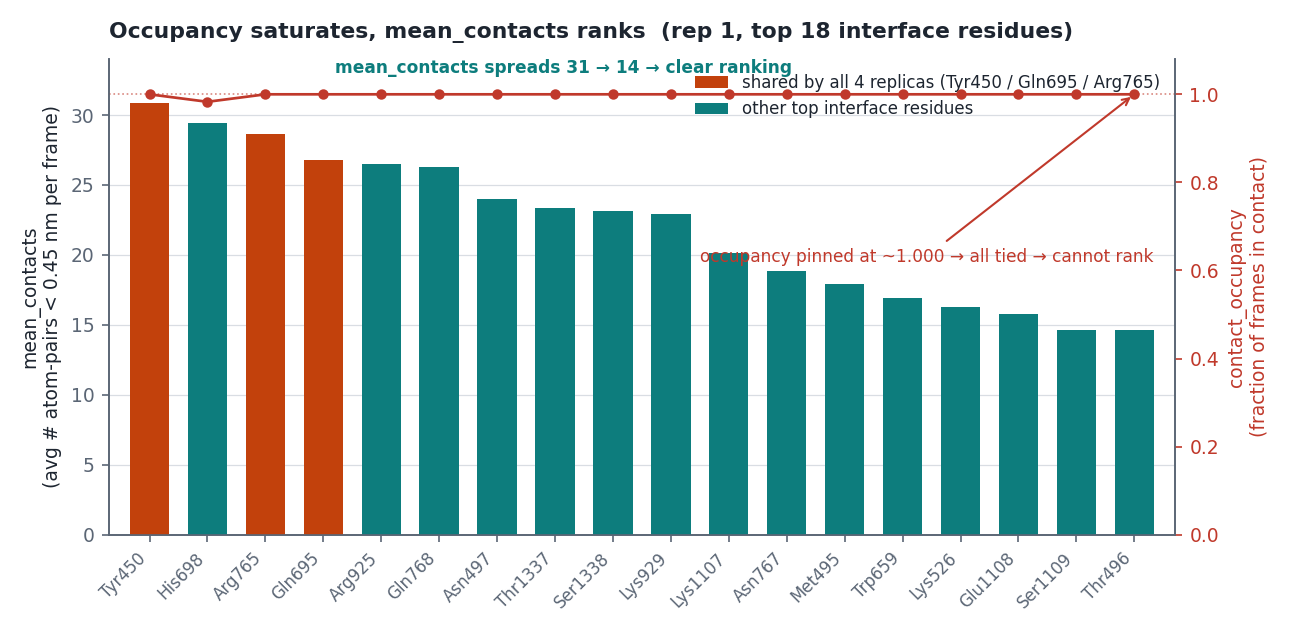
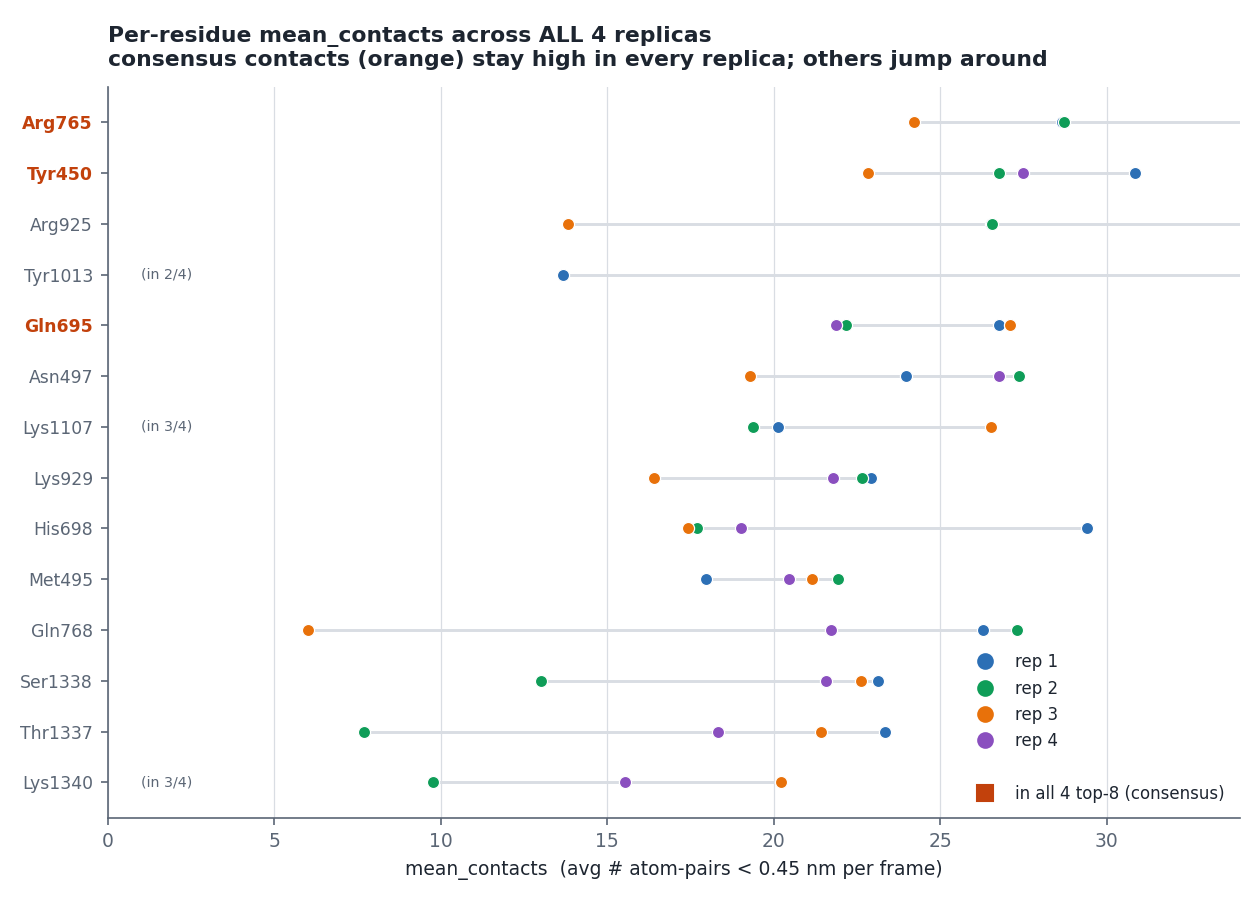
tgt + non − total = 0.000000(四條全部)——它們是總接觸數的精確分割,而且跨 replica r = −0.898(強烈反相關)。所以:tgt − non 的 CV 高達 134.6%,在 n=4 下等於擲硬幣,不能當結論;鹽橋/總接觸 的 CV 是 7.5%,比原始計數的 9.5% 好,是零成本的改善。
為什麼對方難拒絕:pre-register 是攤開假設的好習慣;側枝是加法、可選、只跑少數候選(成本可控、不動主流程)。而且把對話錨定在你最強的主場——free energy、barrier、MD、MSM。你是來「貢獻缺的那塊物理」的合作者,不是來挑錯的。
附帶:這個「binding vs kinetics 的 scoring-axis」在 AI-Protein-Interface dashboard 裡已被登記成一個待裁決的 open decision(D-006/H-011,待 T-011)——你開這話題,是接一個已經在桌上的問題。